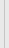菌群多样性组成谱全长分析
菌群组成谱全长测序研究
基于PacBio三代测序平台,能够轻而易举地读取群落微生态系统中所有微生物的rRNA基因/ITS全长序列,不再受制于短序列的局限性,并充分保障全长序列的测序精确性,从而在种甚至菌株等精细水平更全面更深入地解析菌群多样性和组成谱。
产品特色
l 深入获取微生物群落的精细组成信息;
l 预测微生物群落的潜在代谢功能,并指导后续宏基因组测序研究;
l 通过“全微生物组关联研究(Microbiome-Wide Association Studies,MWAS)”,精准鉴定与表型/组间差异相关的关键物种。
应用领域
l 微生物组与环境互作关系的研究;
l 微生物组与宿主共生关系的研究;
l 微生物组在临床医学和精准医疗领域的应用;
l 微生物组在发酵工艺、食品加工/检测等行业的应用。
测序优势
l 自动化、专业、完善的DNA提取扩增体系和样本前处理流程;
l 拥有PacBio RSII和Sequel两种单分子实时测序平台,提供一站式测序服务;
l Greengenes/Silva/HOMD/RDP/Unite/MaarjAM等多个物种注释数据库,更优化物种的分类鉴定;
l 专家教授全方位支持,结果直接用于发表,可按需个性化定制;
l 项目经验丰富全面,涵盖水样、活性污泥、土壤、排泄物、体液、组织等各类样本。
PacBio和Illumina平台对菌群16S rRNA基因测序结果的比较,三代测序的结果能显著提升种水平的物种检测精确性,并大幅改善物种分类信息的不确定性。
送样要求
|
样本 |
样本要求 |
|
土壤类 |
500 mg |
|
排泄物类 |
固体500 mg,液体200 ml |
|
水样类 |
500 ml液体过滤的滤膜 |
|
组织 |
200 mg |
|
微生物基因组总DNA |
体积> 20 ml;浓度> 20 ng/ml |
送样原则:新鲜、足量,请提供三次实验所需用量。
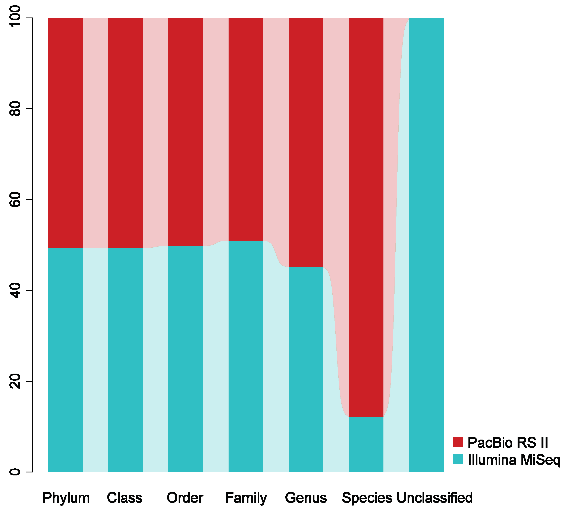
菌群组成谱全长测序分析流程
生物信息分析内容
|
分析项目 |
类别 |
分析要求 |
是否分析 |
|
原始测序数据的质控 |
|
1. 原始测序数据的处理 |
A |
|
ü |
|
2. 疑问序列的剔除 |
A |
|
ü |
|
3. 序列长度分布统计 |
A |
|
ü |
| |
|
|
序列归并和OTU划分 |
|
1. OTU划分 |
B |
|
ü |
|
2. OTU分类地位鉴定 |
B |
|
ü |
|
3. OTU精简和分类鉴定结果统计 |
B |
|
ü |
|
4. 共有OTU的Venn图分析 |
B |
样本(组)≥ 2且≤ 5 |
ü |
| |
|
|
Alpha多样性分析 |
|
1. Rarefaction稀疏曲线 |
B |
|
ü |
|
2. Specaccum物种累积曲线 |
B |
样本≥ 10 |
ü |
|
3. 丰度等级曲线 |
B |
|
ü |
|
4. Alpha多样性指数计算 |
B |
|
ü |
| |
|
|
分类组成分析 |
|
1. 各分类水平的微生物类群数统计 |
B |
|
ü |
|
2. 各分类水平的分类学组成分析 |
B |
|
ü |
|
3. Metastats分析 |
C |
样本(组)≥ 2 |
ü |
|
4. LEfSe分析 |
C |
分组≥ 2 |
ü |
| |
|
|
菌群组成交互式可视化 |
|
1. 系统发育树构建 |
B |
|
ü |
|
2. MEGAN可视化展示 |
B |
|
ü |
|
3. GraPhlAn可视化展示 |
B |
|
ü |
|
4. Krona交互式展示 |
B |
|
ü |
|
5. 热图分析 |
B |
样本≥ 3 |
ü |
| |
|
|
Beta多样性分析 |
|
1. PCA分析 |
B |
样本≥ 3 |
ü |
|
2. UniFrac-PCoA分析 |
B |
样本≥ 3 |
ü |
|
3. UniFrac-MDS分析 |
B |
样本≥ 5 |
ü |
|
4. UniFrac-UPGMA聚类分析 |
B |
样本≥ 3 |
ü |
|
5. UniFrac距离组间/组内差异比较分析 |
B |
分组≥ 2 |
ü |
|
6. 三元相图分析 |
B |
样本(组)= 3 |
ü |
| |
|
|
菌群比较分析和关键物种筛选 |
|
1. PLS-DA分析 |
C |
分组≥ 2 |
ü |
|
2. Adonis/PERMANOVA分析 |
C |
样本≥ 3,并提供影响因素数值;或分组≥ 2 |
ü |
|
3. ANOSIM分析 |
C |
分组≥ 2 |
ü |
|
4. 随机森林分析 |
C |
分组≥ 2 |
ü |
| |
|
|
关联网络分析 |
|
1. 优势物种互作Spearman关联网络分析 |
C |
样本≥ 3 |
ü |
| |
|
|
菌群代谢功能预测 |
|
1. PICRUSt功能预测分析 |
C |
仅限16S rRNA基因数据 |
ü |
|
2. 功能类群分布柱状图分析 |
C |
无分组 |
ü |
|
3. 功能类群分布小提琴图分析 |
C |
分组≥ 2 |
ü |
|
4. 共有功能类群的Venn图分析 |
C |
样本(组)≥ 2且≤ 5 |
ü |
|
5. 结合聚类分析的功能类群热图分析 |
C |
样本≥ 3 |
ü |
| |
|
|
可选性深度分析项目(默认不分析) |
|
1. RDA分析 |
D |
样本≥ 3,并提供影响因素数值;
或分组≥ 2;
默认不分析 |
|
|
2. CAP分析 |
D |
分组≥ 2,默认不分析 |
|
|
3. Fisher’s精确检验 |
D |
样本(组)≥ 2,默认不分析 |
|
|
4. Welch’s t检验 |
D |
分组≥ 2,默认不分析 |
|
|
5. Wilcoxon秩和检验 |
D |
分组≥ 2,默认不分析 |
|
|
6. ANOVA方差分析/Kruskal-Wallis H检验 |
D |
分组≥ 2,默认不分析 |
|
|
7. SparCC关联网络分析 |
D |
样本≥ 5,默认不分析 |
|
|
8. OTU—样本二分网络 |
D |
样本≥ 3,默认不分析 |
|
A:基本信息分析项目;B:常规信息分析项目;C:高级信息分析项目;D:深度信息分析项目(单独收费),划ü的为免费分析项目。
注:默认只提供1份分析报告,分组类型不超过3种。
菌群多样性组成谱全长分析 ,PacBio第三代全长测序常用引物:
细菌16S rDNA:
(1) 对16S rDNA的全长进行PCR扩增
(2) 以扩增产物为模板进行PacBio测序文库的制备
(3) PacBio系统测序16S rDNA的全长序列
(4) 引物信息:27F AGAGTTTGATCMTGGCTCAG
1492R ACCTTGTTACGACTT
古菌16S rDNA:
(1) 对16S rDNA的全长进行PCR扩增
(2) 以扩增产物为模板进行PacBio测序文库的制备
(3) PacBio系统测序16S rDNA的全长序列
(4) 引物信息:21F TTCCGGTTGATCCYGCCGGA
1492R ACCTTGTTACGACTT
真菌ITS全长:
(1) 对真菌ITS的全长进行PCR扩增
(2) 以扩增产物为模板进行PacBio测序文库的制备
(3) PacBio系统测序真菌ITS的全长序列
(4) 引物信息:ITS1F CTTGGTCATTTAGAGGAAGTAA
LR3 CCGTGTTTCAAGACGGG
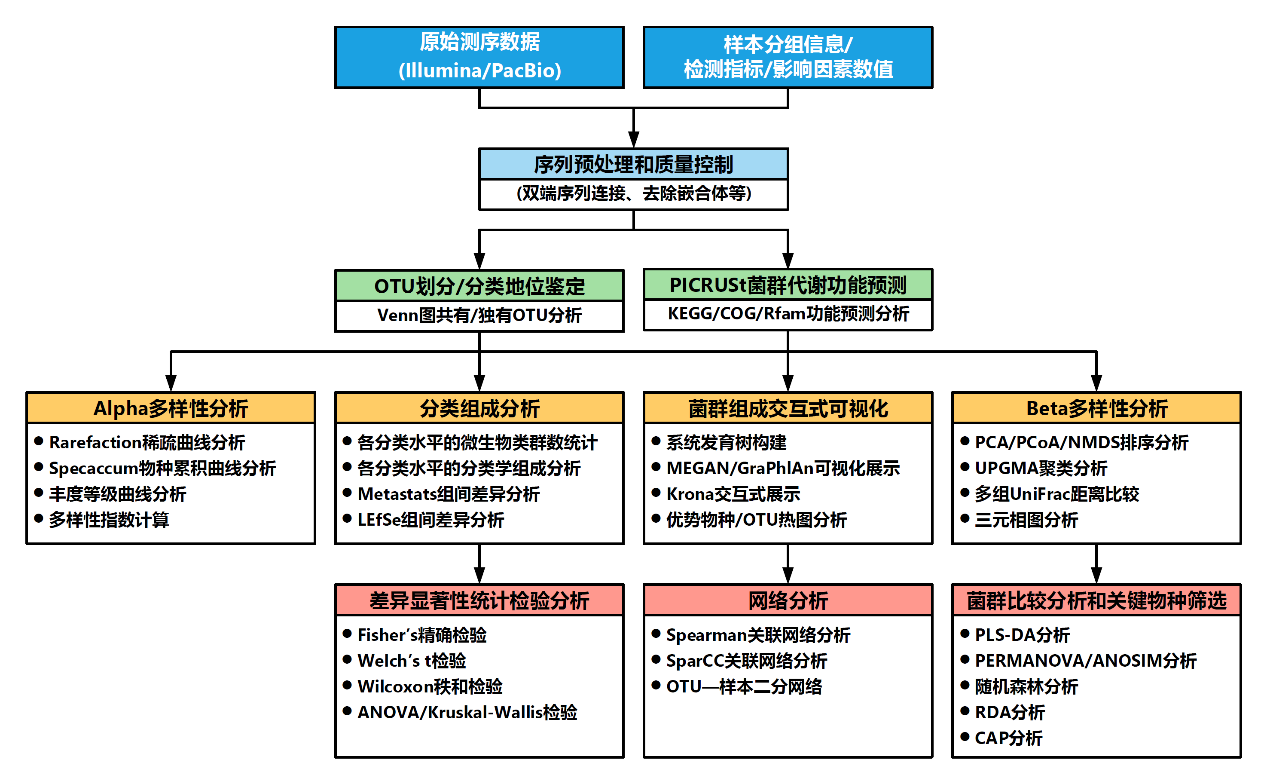
特色分析内容举例
种水平的精细组成图
上图中,横坐标依据样本名排列,每一个柱形图代表一个样本,并以颜色区分各分类单元,纵坐标代表各分类单元的相对丰度,柱子越长,该分类单元在对应样本中的相对丰度越高。
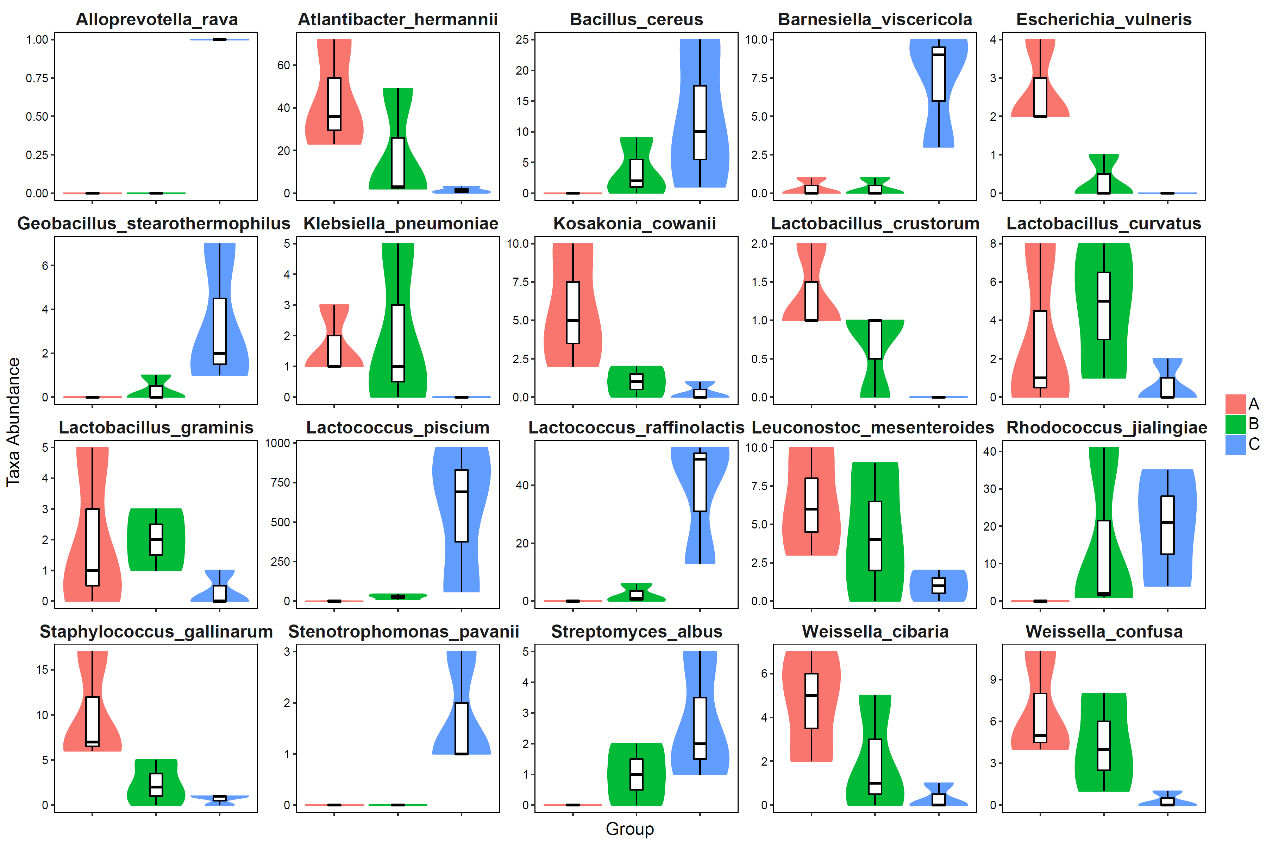
组间差异显著的种水平分类单元的丰度分布小提琴图
上图中,横坐标为组间差异显著的种水平分类单元,纵坐标为各分类单元在各样本(组)内的序列量,以小提琴图结合箱线图的形式展示:其中,小提琴图可以直观地显示数据的分布特征,“小提琴”的“胖瘦”反映了样本数据分布的密度高低(宽度越宽,表明该序列量水平所对应的样本越多);箱线图边框代表上下四分位数间距(Interquartile range,IQR),横线代表中位值,上下触须分别代表上下四分位以外的1.5倍IQR范围,符号“•”表示超过范围的极端值。
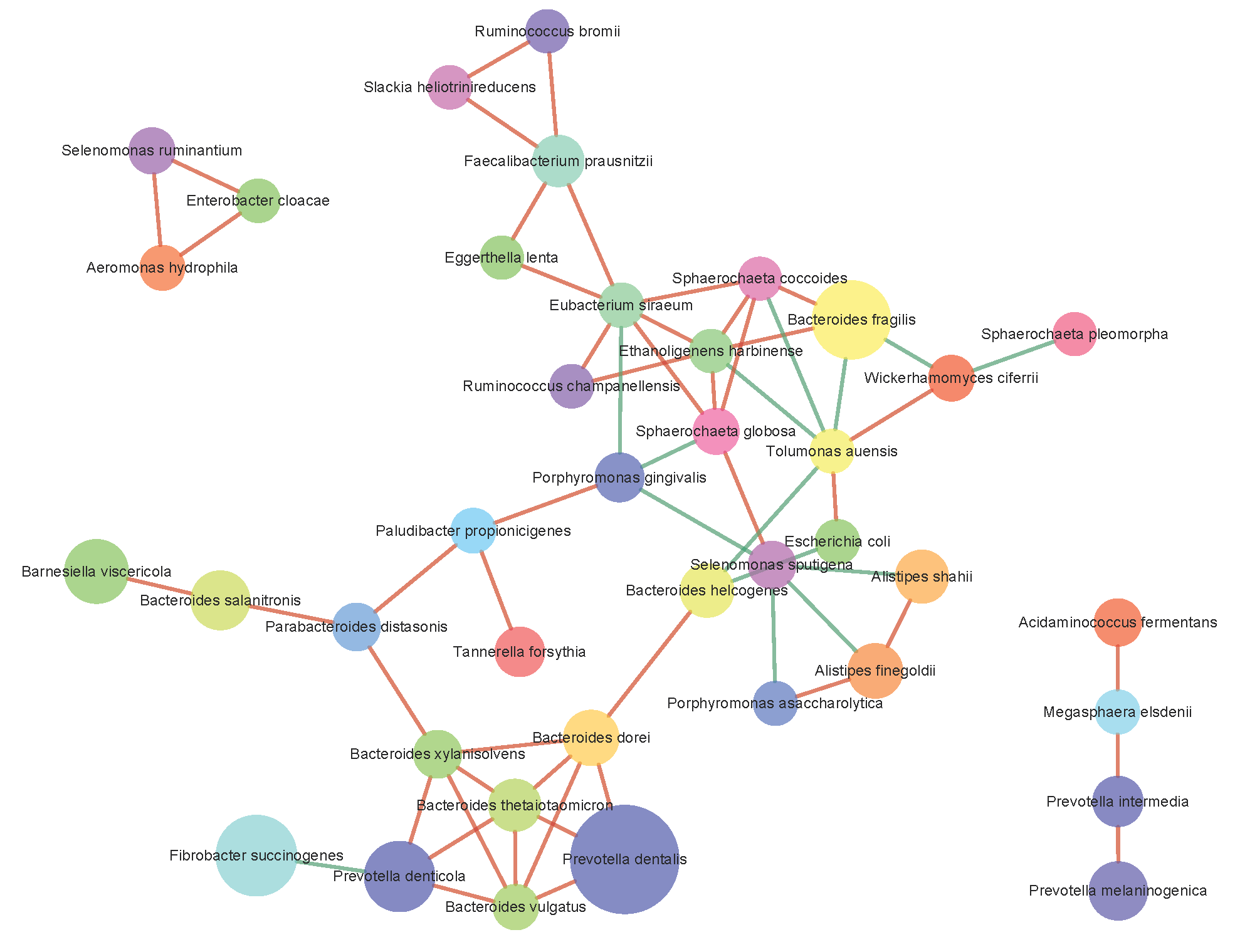
种水平关联网络分析图
上图中,各节点的圆圈代表种水平的各分类单元,以不同的颜色标识,节点之间的连接表明两个物种之间存在相关性,红线表明正相关,绿线表明负相关。通过某节点的连接越多,表明该物种与菌群中其它成员的关联越密切。
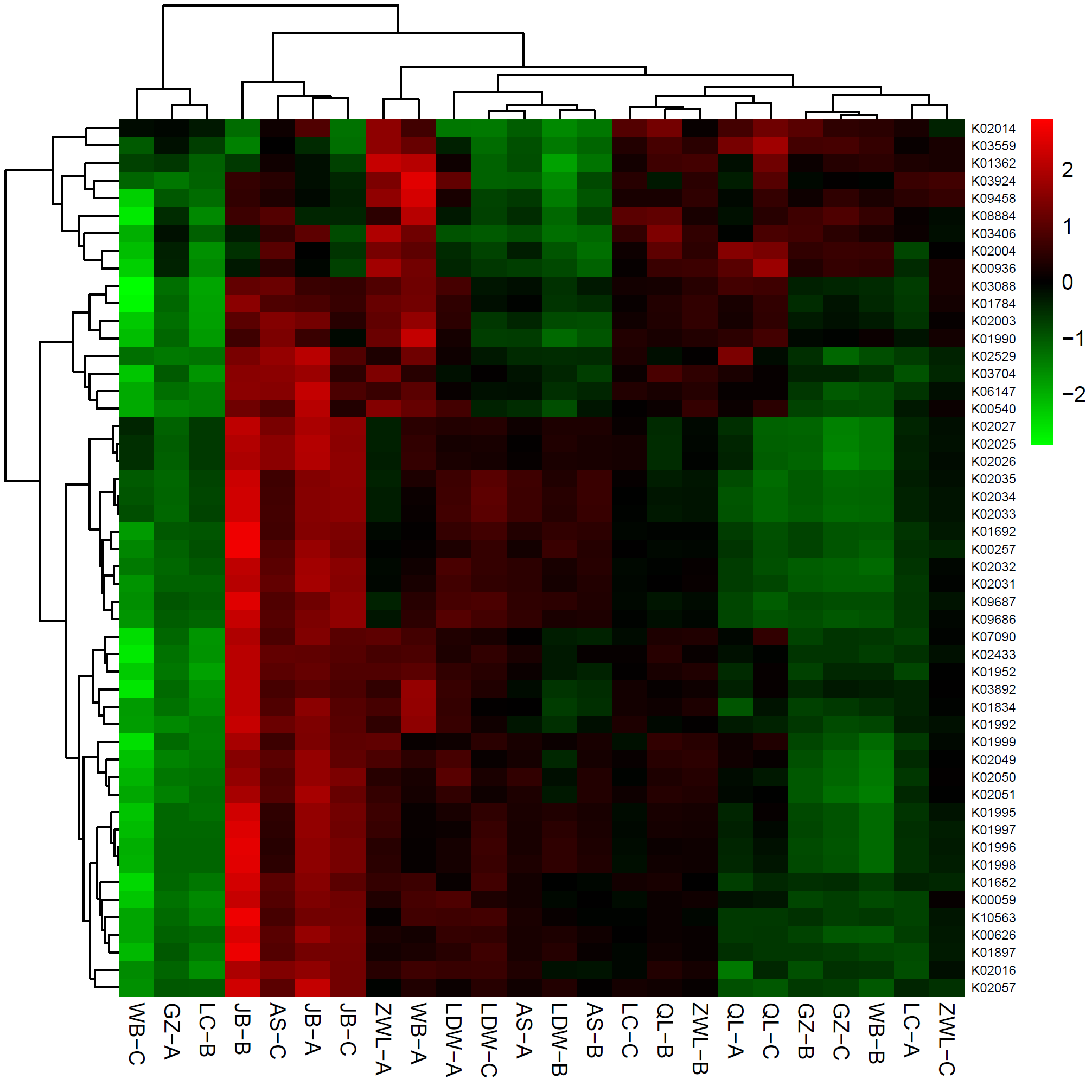
菌群代谢功能预测的KEGG直系同源基因簇(KO)丰度热图
上图中,样本先按照彼此之间功能类群丰度分布的相似度进行聚类,根据聚类结果横向依次排列。同理,功能类群也按照彼此在不同样本中分布的相似度进行聚类,根据聚类结果纵向依次排列。图中,红色代表在对应样本中丰度较高的功能类群,绿色代表丰度较低的功能类群。 |